我们的服务
概览
根据国家药监局要求,医疗器械申请注册证需按照风险等级进行分类。低风险的Ⅰ类医疗器械需要申请备案,高风险的Ⅱ、Ⅲ类医疗器械需要按照国家药监局要求进行注册,同时还需进行临床评估,是否需要进行临床试验则视具体情况而定。
-一类医疗器械备案
-二、三类医疗器械首次注册
-二、三类医疗器械延续注册
-二、三类医疗器械证书注册变更
-注册文件准备与审核
-技术要求起草支持
-产品测试
-内审咨询协助和指导
-补充材料准备指导
-临床评价策略支持
概览
为了在医疗器械注册过程中和注册后满足国家药监局的合规要求,医疗器械制造商应当对政府检查(如飞行检查、内部系统审计等)做出合规性承诺。
-医疗器械注册初期的现场检查提前模拟评估
-制造商取得目标医疗器械市场许可后的突击模拟检查
-内部系统审计的咨询协助与指导
概览
根据GMP / GSP要求,企业应当结合产品特性建立和完善与生产、经营医疗器械相匹配的质量管理制度,确保质量管理体系有效运行。为避免医疗器械质量风险,企业应建立以硬件为基础、文件体系为支撑以及专业人员为保障的质量管理制度,生产质量合规的医疗器械产品。
-医疗器械洁净厂房设施的建立与验证
-医疗产品设计与开发流程咨询服务
-质量管理体系文件的编制和改进咨询服务
-常用设备验证咨询辅导
-设备验证及特殊过程咨询服务
-硬件质量体系合规审核支持
-建立和执行质量体系的培训支持
-支持质量体系评估材料的准备与提交,直至评估通过并成功获得评估报告
概览
MAH注册人制度,即国内医疗器械注册上市许可持有人。除拥有生产许可的医疗器械制造商外,注册人制度为独立医疗机构和医疗器械科研人员在不符合先前要求的持有生产许可的强制性资格的情况下申请上市许可证书提供了合规性。 MAH旨在将营销许可和制造许可解绑,通过让更多研发驱动的医疗机构和没有制造能力的人员加速中国国内的医疗器械创新,使患者受益,使医疗器械研究人员和制造商分别专注于各自的领域。 生产企业按照注册人的委托要求生产,注册人对医疗器械的研发、临床试验、制造、营销和分销等医疗器械的全质量生命周期负责。
-针对质量体系和风险体系,在注册过程中为注册人提供专业的多维度协助
-及时向客户通报更新不同地区注册人制度的最新规定
-协助为注册人匹配合适的制造商
-注册人规划,及双边研发和制造商的对接沟通
-模拟GMP检查以进一步确保审核成功
-为精选供应商提供可靠服务,从委托制造商到产品测试
概览
MAH注册人制度,即国内医疗器械注册上市许可持有人。除拥有生产许可的医疗器械制造商外,注册人制度为独立医疗机构和医疗器械科研人员在不符合先前要求的持有生产许可的强制性资格的情况下申请上市许可证书提供了合规性。 MAH旨在将营销许可和制造许可解绑,通过让更多研发驱动的医疗机构和没有制造能力的人员加速中国国内的医疗器械创新,使患者受益,使医疗器械研究人员和制造商分别专注于各自的领域。 生产企业按照注册人的委托要求生产,注册人对医疗器械的研发、临床试验、制造、营销和分销等医疗器械的全质量生命周期负责。
-文件准备
-现场评估
-受理前申请材料审核
-企业整改咨询服务
-获批后通知
概览
随着中国医疗器械审查制度不断完善和医疗行业高速发展,大量创新型医疗器械正在被引入中国,越来越多国际医疗器械制造商开始将业务的战略重心逐步转向中国市场。而对希望进入中国市场的国际医疗器械制造商而言,在中国指定一位可信赖的代理商是满足中国法律法规要求的必经之路。 拥有超过30年历史的Chindex美中互利在医疗器械行业享有盛誉,一直致力于帮助国际医疗器械制造商加强在中国市场的的政策、监管协同,是国际医疗器械企业布局中国市场、开拓中国业务道路上值得信赖的合作伙伴。
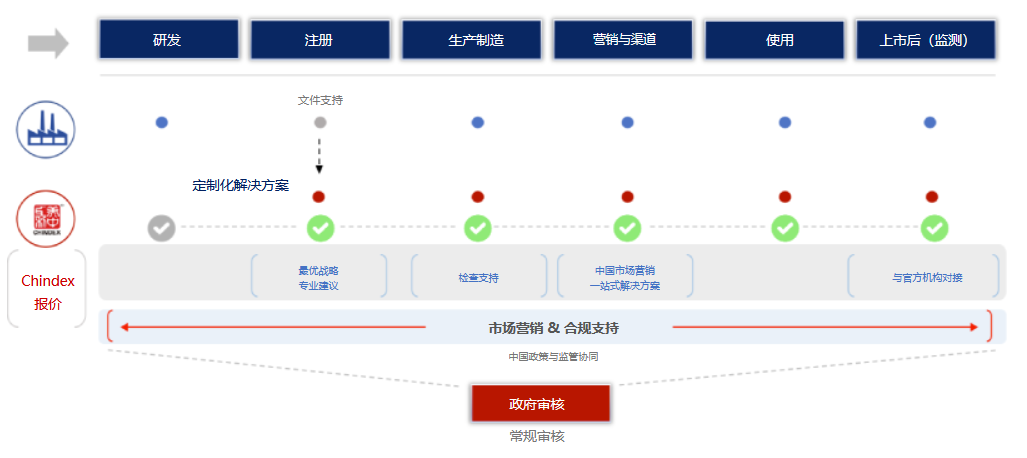
很遗憾,除非制造商在中国设有具有资质的分支机构来处理注册问题,否则这是不合法的且不合规的,进而,海外制造商必须指定中国代理人进行医疗器械注册。
对于NMPA注册费,进口医疗器械第二类医疗器械注册费为21.09万元,第三类医疗器械注册费为30.88万元。此外,还应考虑其他可能的费用,例如文件翻译费、临床试验费等。
注册材料经国家药监局受理后,第二类医疗器械技术审评需要60个工作日,第三类医疗器械技术审评需要90个工作日。国家药监局要求补充材料的,生产企业有义务在一年内提交,否则可能终止评审。
有 CE 或 FDA 证书的医疗器械可以在没有 NMPA 注册证的情况下进入特别行政区。对于大湾区和海南省作为试点地区,在香港或澳门已上市的医疗器械,经当地政府批准,试点地区定点医疗机构愿意采购临床急需医疗器械的,可进入试点地区。
京ICP备11010059号-1  京公网安备11010502035116号 (京)网药械信息备字(2023)第 00034 号 版权所有©澳门新葡萄新京6663
京公网安备11010502035116号 (京)网药械信息备字(2023)第 00034 号 版权所有©澳门新葡萄新京6663
京公网安备11010502035116号

